
Neurofibromatosis Type 1
What Is Neurofibromatosis Type 1?
Neurofibromatosis type 1 is one of the most common neurocutaneous disorders, affecting about 1 in 3000 individuals. It is inherited in an autosomal dominant fashion with near complete penetrance; however, it has variable expression. In half of NF1 cases, patients inherit the mutated NF1 gene from an affected parent. The other cases occur as a result of a mutation in the gene.
The NF1 gene is a tumor suppressor gene localized to chromosome 17 and mostly affects growth of Schwann cell and glial cell derived tissues both in the central and peripheral nervous system. Loss of function or expression due to a mutation leads to increase in cell proliferation resulting in development of glial tumors. Neurofibromas are the most common tumor in NF1 that often present in puberty and increase in number and size with age. Most are benign and rarely undergo malignant transformation.
The underlying cause of seizures in patients with NF1 is not always clear. NF1 cortical lesions have not been clearly found to be associated as an epileptic focus. In addition, the bright objects have not been found to be associated with focal EEG findings in patients with NF1. However, it has been suggested that a higher number of NBOs is associated with a higher risk for seizures. However, this does not eliminate that the cause of epilepsy might be genetic and probably related to NF1. More commonly, intracranial tumors seen in NF1 patients have been found to directly correlate with epilepsy.
Common clinical findings on exam include café au lait spots, neurofibromas, Lisch nodules, and optic gliomas. MRI brain imaging may show T2 signal change in the subcortical region in addition to tumors, often call neurofibroma bright objects (NBOs).
Other presenting symptoms may include cognitive dysfunction, pain in specific nerve distribution (usually due to presence of neurofibroma), seizures, changes in vision that may be related to optic gliomas, seizures from intracranial tumors, stenosis of major intracranial arteries leading to Moyamoya phenomenon, headaches, and progressive neurologic deficits.
Epilepsy is seen in about 4 to 7% of patients with NF1—double the risk factor compared to the general population. The onset of epilepsy can occur at any age and effects both genders equally.
Learn More:
Contact Our HelplineWhat Types of Seizures Are Seen?
Different seizure types and syndromes have been described in NF1. These include generalized onset seizures (tonic-clonic and atonic), as well as focal (partial) onset seizures with and without secondary generalization. Overall, the most common is focal onset seizures with or without secondary generalization. There is a higher prevalence of West syndrome (infantile spasms) and febrile seizures in patients with NF1 than the general population.
How Is Neurofibromatosis Type 1 Diagnoised?
EEGs are abnormal in about 25% of patients with NF1. The EEG findings can vary from normal to focal or multifocal spike waves, spike and slow spike wave complexes at 2 Hz consistent with Lennox-Gastaut syndrome, and even a hypsarrhythmia background. Focal abnormalities on EEG are the most common.
There are seven hallmarks of NF1 and two or more are needed to make the diagnosis:
- At least six café-au-lait macules more than 5 mm in greater diameter in pre-pubertal children, and more than 15mm in greatest diameter in post-pubertal children
- Axillary and/or inguinal freckling (usually present between ages 3 and 5 years)
- At least 2 Lisch nodules
- Neurofibromas: more than one cutaneous neurofibroma or one plexiform neurofibroma
- Characteristic bone anomalies, such as a sphenoid dysplasia or thinning of long bone cortex, with or without pseudoarthrosis
- Optic nerve glioma
- First-degree relative with neurofibromatosis type 1
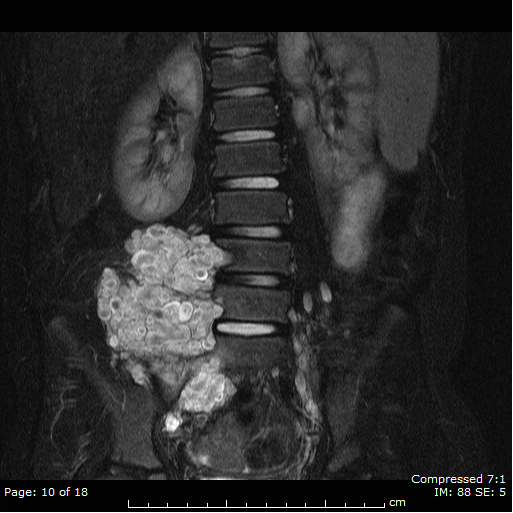
Neurofibromatosis Type 1
Plexiform Neurofibroma
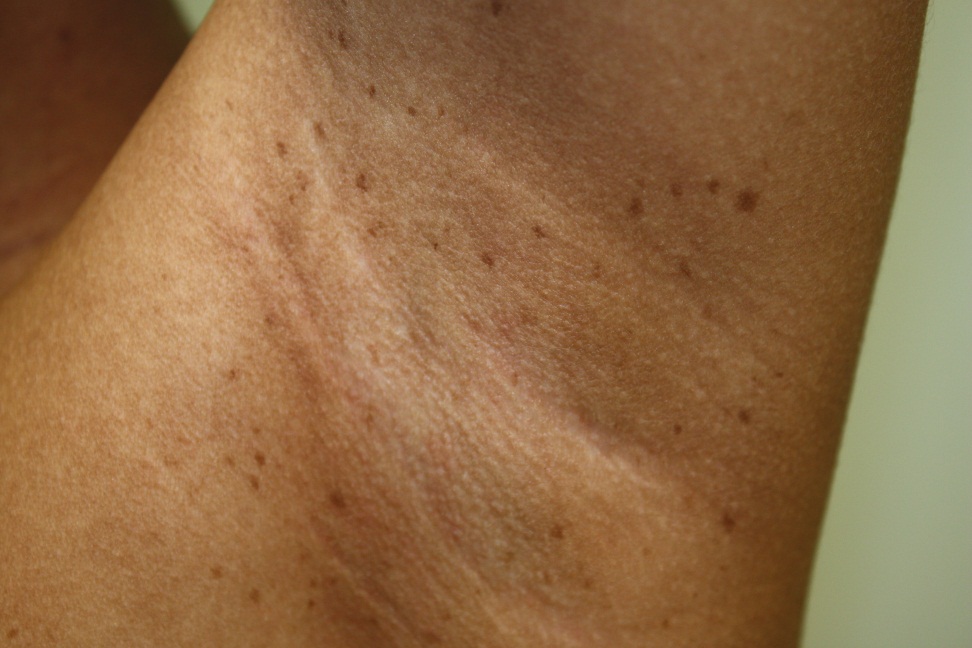
Neurofibromatosis Type 1
Axillary Freckling
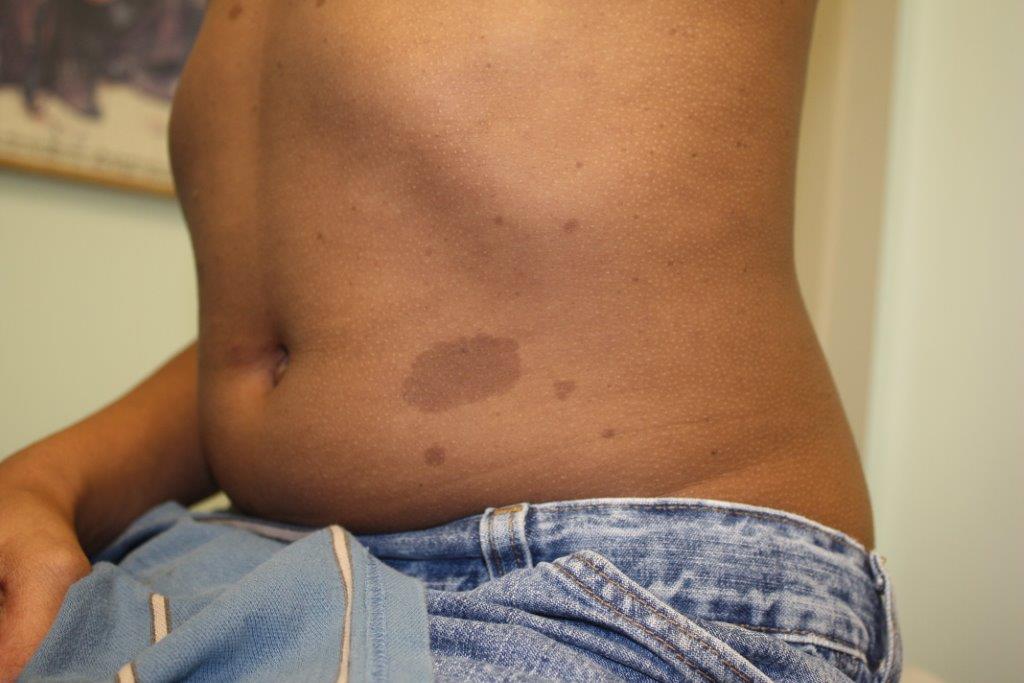
Neurofibromatosis Type 1
Café au Lait Macule
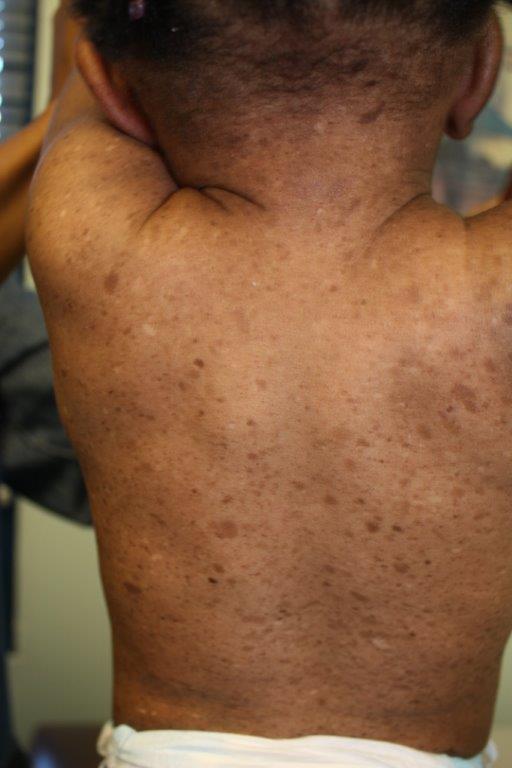
Neurofibromatosis Type 1
Café au Lait Macule
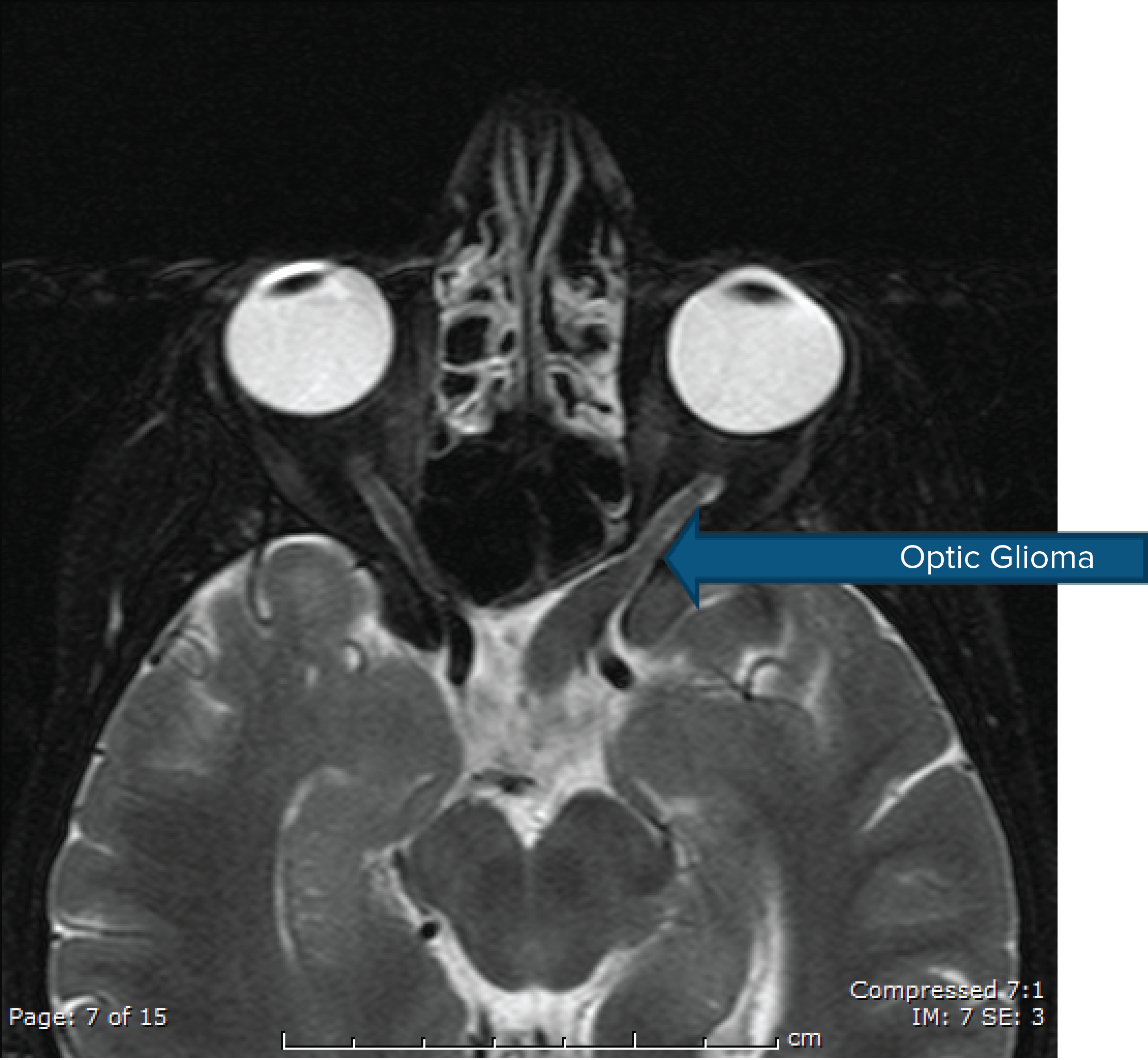
Neurofibromatosis Type 1
Optic Glioma
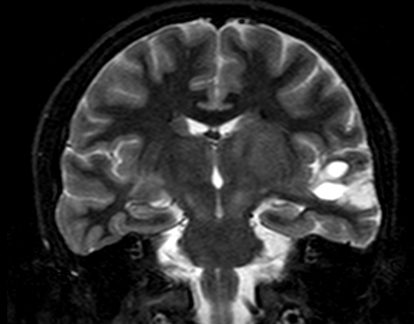
Neurofibroma bright objects
with cystic changes noted on MRI
(T2 weighted imaging) in a patient
with refractory epilepsy
Learn More:
Find Your Local Epilepsy FoundationHow Is Neurofibromatosis Type 1 Treated?
Treatment is symptomatic and supportive as there is no known cure.
- Counseling and anticipatory guidance and surveillance for treatable complications, which includes blood pressure screening, growth, pubertal status, and ophthalmologic examinations.
- Regular neuropsychological evaluations should also be done especially in children.
- Repeat imaging is usually done on a clinical basis as the lesions are slow growing and are usually not treatable. Malignant tumors are managed with appropriate neurosurgical measures, chemotherapy, and radiation.
For epilepsy, treatment involves anti-seizure medications, but no specific medication is favored. Some patients may become refractory to medical management. Overall patients with NF1 and seizures are generally more difficult to treat with a single agent and are more likely to be refractory. If the cause of seizures is due to an intracranial tumor, if appropriate, epilepsy surgery may be recommended.
What Is the Outlook?
The outlook for patients with NF1 is relatively good if the degree of symptoms is mild to moderate and many patients live healthy lives. A common comorbidity of patients with NF1 is the psycho-social concerns that include cognitive disabilities and social interactions. Patients with NF1 have a higher risk for cognitive dysfunction if they have epilepsy.
In the more severe cases of NF1, there can be more severe and disabling symptoms due to pain, significant learning disabilities, psychological problems, refractory epilepsy, and cosmetic difficulties.
Learn More:
Donate to Support Our MissionResources
Epilepsy Centers
Epilepsy centers provide you with a team of specialists to help you diagnose your epilepsy and explore treatment options.
Epilepsy Medication
Find in-depth information on anti-seizure medications so you know what to ask your doctor.
Epilepsy and Seizures Helpline
Call our Epilepsy and Seizures Helpline and talk with an epilepsy information specialist or submit a question online.
Tools & Resources
Get information, tips, and more to help you manage your epilepsy.
Related Stories


Press Releases
New Joint Statement Highlights Seizure Detection Devices as Tool for Safety in Epilepsy Care
Read Story Press Releases
Epilepsy Foundation of America Awards $250,000 at 14th Annual Shark Tank Competition
Read Story