Myoclonic Atonic Epilepsy Doose Syndrome

What is myoclonic atonic epilepsy (Doose Syndrome)?
Myoclonic atonic epilepsy (MAE), typically known as Doose syndrome, was first described by Dr. Herman Doose from Germany in 1970. It is an uncommon childhood epilepsy syndrome that accounts for 1 to 2 out of 100 (1 to 2%) of all childhood-onset epilepsies.
- Usually the first seizure occurs between 2 and 6 years of age.
- Boys tend to be affected more than girls (two thirds to three quarters of children with MAE will be boys).
- More than half of children start with a generalized tonic-clonic seizure with or without fever. Within days to months, drop seizures begin. Many children also develop myoclonic jerks and absence seizures, and some may develop tonic seizures. Most children with MAE are developing normally when seizures begin.
Is myoclonic atonic epilepsy genetic or inherited?
Genetics play an important role in this disorder. About 1 out of 3 families have a history of seizures.
While most children do not have an identified gene mutation, mutations in various genes, including SCN1A, SCN1B, SCN2A, SLC2A1, CHD2, SYNGAP1 and KIAA2022, have been identified in some cases. This epilepsy syndrome is considered part of a group of genetic epilepsies with febrile seizures plus (GEFS+).
What do the seizures in myoclonic atonic epilepsy look like?
Children with MAE may have multiple seizure types:
- Drop seizures, characterized by an abrupt fall with loss of body tone (atonic) or by a brief body jerk followed by an abrupt fall (myoclonic atonic), occur in everyone.
- Generalized tonic-clonic seizures are seen in most children. These may be triggered by fever.
- Absence seizures and myoclonic seizures are seen in many cases.
- Tonic seizures can be seen later in the course and often are predictive of poorer outcomes.
Though all seizure types can result in status epilepticus, about 1 in 3 children with MAE have episodes of non-convulsive status epilepticus. Seizures tend to occur more often early in the morning than during the day.
How is myoclonic atonic epilepsy diagnosed?
MAE is diagnosed based on the description of the seizures, with myoclonic atonic seizures being the most common type. Information also comes from tests such as:
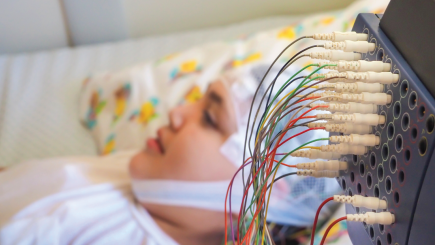
- EEG (electroencephalogram): children with MAE may have normal EEGs at first, but most have abnormal EEGs showing frequent generalized spike-wave activity in brief bursts of 2 to 5 Hz. Background may be normal or abnormal. (See the photo at the top of the page.)
- MRI (magnetic resonance imaging) of the brain is normal.
How Is Myoclonic Atonic Epilepsy Treated?
Medications
Seizures in children with MAE are often difficult to treat and may not respond well to medication. Medications are chosen based on the seizure types:
- Generalized tonic-clonic, myoclonic, and myoclonic atonic seizures are typically treated with valproic acid/divalproex, lamotrigine, levetiracetam, topiramate, zonisamide, rufinamide, clobazam, and felbamate in the United States.
- Absence seizures are typically treated with ethosuximide, valproic acid/divalproex, or lamotrigine.
Some seizure medications should not be used in children with MAE because they can worsen certain seizure types. These include carbamazepine, oxcarbazepine, phenytoin, and vigabatrin.
Non-medication Therapies
The ketogenic diet is often very effective for MAE and should be considered early. Vagus nerve stimulation (VNS) has been used, but some reports have not shown benefit.
Rescue Therapies
Children with MAE who have long or clusters of seizures may need emergency medical treatment or treatment with a rescue therapy. Examples of rescue therapies may include diazepam rectal gel (Diastat) or another form of benzodiazepine given into the nose (intranasally) or under the tongue.
- Parents of children with MAE should talk to their treating neurologist or health care provider to learn about seizure emergencies and options for home rescue medication.
- If seizures do not stop with rescue medication, or in cases of suspected non-convulsive status epilepticus (where the child has impaired awareness, sleepiness, unsteadiness, drooling and twitching of the face, arms or legs), the child may need emergency medical care.
What Is The Outlook For Children With Myoclonic Atonic Epilepsy?
The prognosis for children with MAE can vary.
- While seizures often respond poorly to medication, about 2 out of 3 children ultimately outgrow their epilepsy and can wean off medication.
- About one third have persisting seizures.
- Development typically improves once seizures are controlled, with some children returning to normal function. Others may be left with varying degrees of intellectual disability.
- Intellectual disability is more severe in children whose seizures do not go away and in those with tonic seizures and recurrent, non-convulsive status epilepticus.
Resources
- Doose Syndrome Epilepsy Alliance, a member of the Rare Epilepsy Network (REN)
- International League Against Epilepsy (ILAE) on epilepsy with myoclonic-atonic seizures
- National Organization for Rare Disorders (NORD) on Doose syndrome
- What’s a clinical trial and why should you join one? Find out here.
- Find a Clinical Trial
- Information on current clinical trials also can be found at www.clinicaltrials.gov.
- Find epilepsy therapies in various stages of development in our Epilepsy Pipeline Tracker
Resources
Epilepsy Centers
Epilepsy centers provide you with a team of specialists to help you diagnose your epilepsy and explore treatment options.
Epilepsy Medication
Find in-depth information on anti-seizure medications so you know what to ask your doctor.
Epilepsy and Seizures 24/7 Helpline
Call our Epilepsy and Seizures 24/7 Helpline and talk with an epilepsy information specialist or submit a question online.
Tools & Resources
Get information, tips, and more to help you manage your epilepsy.
Related Stories

Press Releases
Patient Organizations Applaud Release of Affordable Care Act Rule to Protect All Patients from Discrimination When Accessing Health Care
Read Story