Early Infantile Developmental & Epileptic Encephalopathy
What Is Early Infantile Developmental and Epileptic Encephalopathy (EIDEE)?
Early Infantile DEE is a rare form of neonatal epilepsy occurring in 10 in 100,000 live births. EIDEE was previously known as Ohtahara syndrome or early infantile epileptic encephalopathy (EIEE) and Early Myoclonic Encephalopathy (EME).
Children with EIDEE may have frequent and drug-resistant seizures in the first 3 months of life. They may also have an abnormal neurological exam even before their seizures start and have abnormal EEG findings between seizures. EIDEE affects both boys and girls. Family history, birth, and birth history are often normal for infants with this condition.
What Types of Seizures Are Seen In EIDEE?
The most common seizures include tonic and or myoclonic seizures. Infants could also have other seizure types including:
- Focal tonic
- Generalized tonic
- Focal
- Clonic
Usually, multiple different seizure types are seen. Seizures can often start as one type and then advance into other types.
What Causes EIDEE?
In up to 80% of patients, EIDEE is caused by an underlying structural, genetic, or metabolic reason. Structure issues in the brain may affect both sides of the brain. They can also be focal, which means it can affect only one part of the brain.
A causative genetic change is found in more than half of babies and could involve changes in genes, such as KCNQ2, SCN2A, SCN8A, CDKL5, STXBP1, UBA52 or others.
Metabolic causes can include:
- Non-ketotic hyperglycinemia
- Amino and organic acid disorders
- Urea cycle disorders
- Mitochondrial disorders
- Pyridoxine and pyridoxal-5-phosphate disorders
- Sulfite oxidase deficiency
- Menke syndrome or Zellweger syndrome
How Is EIDEE diagnosed?
Diagnosing an infant with EIDEE syndrome is based on signs and symptoms. It is also based on the baby's EEG results, which is the most important test in diagnosis. The following exams and tests are used:
- Examination is severely abnormal with low tone and posture, abnormal movements, and poor visual interest. Some of these abnormalities could be noted even before seizure onset but can be challenging to recognize due to early seizure onset.
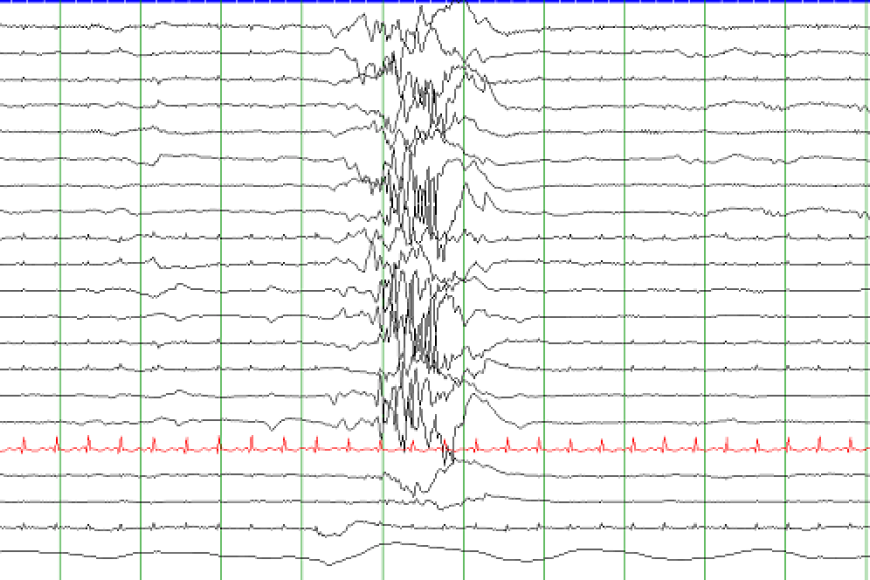
- The EEG is very abnormal (see image at the top of the page) both during sleep and when the infant is awake.
- An MRI (magnetic resonance imaging) scan is required to look for structural changes in the brain that could cause EIDEE. If the MRI is normal, follow up MRIs can show atrophy (shrinkage) of the brain.
- Genetic testing with chromosomal microarray, karyotype, epilepsy gene panel, whole exome or whole genome sequencing should be done in all cases of EIDEE unless another cause is known.
Other tests that could be considered include blood, urine, cerebrospinal fluid (CSF) tests to evaluate for metabolic disorders.
How Is EIDEE Treated?
EIDEE treatment is based on the underlying cause. For example, if a particular chemical problem is found then certain vitamin therapies may be prescribed. If a genetic mutation is found in SCN2A or SCN8A, then certain medications in high doses could be helpful. Treatment options for EIDEE may include:
- Anti-seizure medications
- Ketogenic diet
- Surgery if a focal abnormality is found on an MRI
- Anti-seizure devices
Anti-seizure devices include VNS, RNS, DBS. This treatment is considered, although not typically used in infants. Caregivers should consider discussing newer precision therapies for certain cases. This type of therapy is unique to the person using them. For example, precision therapy for a person with EIDEE will factor in the root cause of their condition to help better treat their symptoms. Additionally, patients usually need multidisciplinary care as they can have orthopedic problems, feeding issues, and vision impairment.
What Is the Outlook for People With EIDEE?
The outlook for infants with EIDEE is often poor. Many children progress to have infantile epileptic spasms syndrome (West Syndrome) or Lennox-Gastaut syndrome (LGS). Some children with EIDEE syndrome may die early in life usually due to lung infections related to severe disability. Those who survive are typically left with severe physical and cognitive disabilities. Children who are candidates for epilepsy surgery should be considered early, as this may lead to improved seizure control and developmental outcome.
Hope for children with EIDEE relies on the medical and research communities understanding more about the causes and what treatments may work best. The Rare Epilepsy Network (REN) connects families with research in these areas.
Resources
Epilepsy Centers
Epilepsy centers provide you with a team of specialists to help you diagnose your epilepsy and explore treatment options.
Epilepsy Medication
Find in-depth information on anti-seizure medications so you know what to ask your doctor.
Epilepsy and Seizures 24/7 Helpline
Call our Epilepsy and Seizures 24/7 Helpline and talk with an epilepsy information specialist or submit a question online.
Tools & Resources
Get information, tips, and more to help you manage your epilepsy.
Related Stories