Lennox Gastaut Syndrome LGS
"This Is LGS" - Brought to you by LGS Together
What is Lennox-Gastaut syndrome?
- The Lennox-Gastaut syndrome (LGS) is a type of epilepsy with multiple different types of seizures, particularly tonic (stiffening) and atonic (drop) seizures.
- Intellectual development is usually delayed and often worsens over time. Behavioral problems, including hyperactivity, agitation, aggression and autism, are common.
- Lennox-Gastaut syndrome is a type of “epileptic encephalopathy.” This terms means that the frequent seizures and very abnormal EEG (electroencephalograph) activity worsens cognitive and behavioral problems.
- The cause of the disorder is unknown in 1 out of 4 children.
Learn More:
Contact Our HelplineHow common is it?
"Lennox-Gastaut syndrome accounts for only 2 to 5% of childhood epilepsies."
Yet children with this type of epilepsy are well-known to both pediatric and adult neurologists. Seizures in people with LGS are hard to control and continue throughout life. The intellectual and behavioral problems seen with LGS add to difficulties in managing life.
Although seizures often begin by early preschool years, it typically takes time for all the clinical and EEG features to appear.

- Diagnosis may not be obvious when seizures begin. It may take several years to diagnosis LGS.
- Some young children have the types of seizures typical for LGS but don’t have all features of LGS. These children need close follow-up to watch for other signs of LGS.
- LGS beginning after age 10 is not common.
In many cases, Lennox-Gastaut syndrome develops after another type of epilepsy, such as West syndrome (infantile spasms), Ohtahara syndrome or Epilepsy in Infancy with Migrating Focal Seizures.
Most children have developmental or intellectual problems before LGS is diagnosed.
LGS is slightly more common in boys. No racial differences have been documented.
How Is It Diagnosed?
For a child or adult to be diagnosed with Lennox-Gastaut syndrom, three key features must be present. These include the following.
Multiple Seizure Types
- Seizure types in LGS include tonic, atonic or drop attacks, atypical absence, myoclonic, and generalized tonic-clonic seizures starting in childhood.
- Seizures begin in childhood but different types may emerge over time. Focal (one area of the brain) seizures become more common in teens and adults.
A Characteristic EEG Pattern
The EEG during wakefulness shows diffuse or widespread background slowing and slow spike-wave bursts. In sleep, a characteristic pattern, termed “generalized paroxysmal fast activity” is seen. This may sometimes correlate with subtle tonic activity.
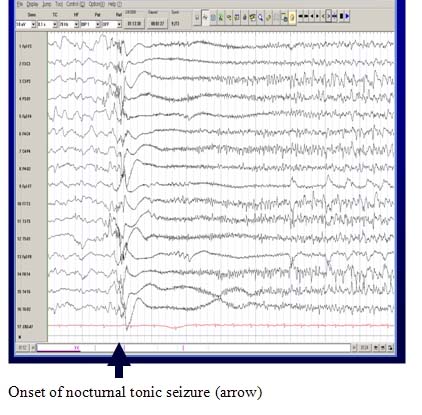
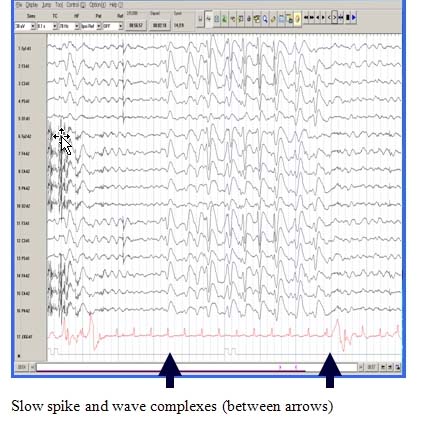
Cognitive Impairment, Behavior Problems or Developmental Delay
Prior to seizure onset, 70-80% (or 7 to 8 out of 10) of children have a history of delayed development and neurological problems. When seizures begin, these problems are almost always seen and often get worse over time.
What causes LGS?
There are many possible causes of Lennox-Gastaut syndrome. The cause is eventually found in over 75% (3 out of 4) of people.
"There are 3 main groups of known causes."
Structural Causes
Structural causes are divided into two main groups:
- Congenital Brain Malformations: This term means that part of the brain has developed abnormally, making it more prone to seizures.
- The abnormal brain development may be caused by a genetic abnormality, an infection that occurred early during pregnancy, or some type of injury that occurred early in pregnancy.
- Often the cause of the abnormal brain development is not known.
- When a child is diagnosed with a congenital brain malformation, it is normal for mothers to worry that this may be due to something they did during the pregnancy, such as having a glass of wine, being briefly exposed to a certain chemical, or spending a brief period of time in a Jacuzzi. They should be reassured that these activities do not lead to severe congenital brain malformations.
- Acquired Brain Injury: This means that the brain has developed normally, but a brain injury occurs later. Brain injuries can include lack of adequate oxygen or blood flow, stroke, bleeding, infections such as meningitis or encephalitis, or head injury.
Brain malformations or injuries may affect focal (specific) areas of the brain or may affect the brain more diffusely.
Genetic Disorders
Genetic disorders are increasingly recognized as a cause of early-onset, severe epilepsy such as LGS.
- Sometimes, genetic disorders can lead to structural changes in the brain, such as in Tuberous Sclerosis Complex.
- In other genetic disorders, the brain appears normal on MRI (magnetic resonance imaging).
- The specific genetic disorder can result in other symptoms, such as characteristic facial appearance (seen in Down syndrome) or other neurological symptoms (like movement disorders or developmental problems).
- Many genetic disorders that lead to an early onset epilepsy arise “de novo.” This means that there is no family history of other affected relatives.
Learn about Genetics to Better Understand One Cause of LGS
Metabolic Disorders
Metabolic disorders rarely cause LGS, but some may be associated with specific treatments.
- It’s important to exclude these during the diagnostic work-up with blood and urine tests.
- Many metabolic disorders can be associated with specific changes on brain MRI scans.
How do you treat LGS?
"The first treatment for anyone epilepsy is antiseizure medications."
Antiseizure Medications
The first treatment for any person with epilepsy, including children and adults with Lennox-Gastaut syndrome, is antiseizure medications. Seizures with LGS usually are not controlled with one seizure medicine. Two or more medications are often needed. Even then, medicines may not control seizures enough and the person continues to be at risk for injuries, seizure emergencies, and worsening of cognition (thinking) and behavior.
- Most seizures in LGS don’t respond well to usual seizure medicines, and side effects can be hard to tolerate.
- Partial relief from seizures may be found with medications.
- One of the most common seizure medicines used in LGS is valproic acid.
- Medicines that have proven to reduce seizures in many persons with LGS are lamotrigine, topiramate, felbamate, rufinamide, clobazam, alternative therapies and most recently fenfluramine. However, other medications may also be helpful.
- When seizures don't respond to medicines, it’s time to look at other treatments.
Dietary Therapies
Dietary treatments can often reduce the number of seizures in people with LGS. The number or doses of some seizure medications may be lowered if diet therapy works well.
The dietary therapies used for LGS and other forms of epilepsy include the ketogenic diet, modified Atkins diet, and Low Glycemic Index diet.
- These diets restrict some food choices, so they can be difficult to stay on for long periods.
- The ketogenic diet is most restrictive and is often used when several medications have failed. It can also be easily given in children or people who get their food and nutrition through a tube in their stomach. Parents or other caregivers can control the types and amount of foods given. The ketogenic diet is prescribed by a physician and carefully monitored by a dietitian.
- The modified Atkins diet and low glycemic diet are more liberal and easier to use. These are being tried more in adolescent and adults.
Learn more about dietary therapies so you can discuss this option with your health care provider. Follow our Keto News to stay up to date about dietary therapies.
Surgery and Devices
Rarely, people with Lennox-Gastaut syndrome will be found to have a focal area of brain that can be removed to help control seizures.
- Corpus Callosotomy
- This type of brain surgery can be done to separate some of the connections between the two sides of the brain.
- This surgery has been very helpful to decrease the frequency of drop attacks. Since these are the most frequent seizures in LGS and cause the most injury, corpus callostomy is often considered for people with LGS.
- Vagus Nerve Stimulation (VNS)
- The most common device used in people with Lennox-Gastaut syndrome is vagus nerve stimulation with VNS Therapy®.
- Surgery is needed to place the device in the left chest area and the left side of the neck. It does not require surgery in the brain.
- Repeat surgeries may be needed after a number of years to replace the generator when the battery in it wears down or to replace an electrode wire. This type of surgery is an outpatient procedure that usually takes 1 to 2 hours at most.
- Learn more about VNS so you can discuss this option with your health care team.
Rescue Medications
About 50 to 75% of people with LGS have one or more episodes of non-convulsive status epilepticus.
- This type of seizure emergency consists of ongoing atypical absence seizures.
- The person’s awareness is altered and they may appear confused for long periods.
- They may have frequent to near continuous twitching of the arms or face (myoclonic seizures).
- Atonic symptoms, such as a drop of the head or part of the body, may happen with clusters of brief tonic (stiffening) seizures too.
Convulsive status epilepticus can also occur in people with LGS. This type of seizure emergency happens when a generalized seizure with loss of conscioueness lasts 5 minutes or longer or repeated seizures happen without the person coming back to awareness between seizures.
"Status epilepticus is a seizure emergency."
- It is critical that families and caregivers of children and adults with LGS become familiar with the types of seizures their loved one has. Families should know seizure first aid and have a plan for responding to seizures.
- Rescue therapies, or treatments that can be given to help stop or shorten clusters of seizures, are an important part of seizure care for people with LGS.
Family Support
Families of people with LGS should work closely with a social worker to find resources to maximize quality of life for their child. Respite help is also important for the family.

As the child grows older and ages out of school resources, other help will be needed. The social worker can help explore financial resources, such as social security and health insurance options, vocational or day programs, and residential or assisted living options, if appropriate.
Children with LGS also will need an Individualized Education Plan. Many may benefit from ongoing physical, occupational and speech therapy. It’s important to involve the school nurse and mental health resources early.
"We Can Help You Find Resources and Support"
- Call our 24/7 Helpline at 800-332-1000 (en Español 1-866-748-8008)
- Go to our Helpline webpage to "Ask a Question" online or "Search for Resources"
- Reach out to your local Epilepsy Foundation
Be Prepared When A Seizure Happens
- Know - and teach others - seizure first aid. It's Stay. Safe. Side.
- Fill out - and share - a seizure action plan so others will know what you want them to do for you.
Learn More:
Find Your Local Epilepsy FoundationWhat is the long term outcome?
The long-term prognosis for seizures, learning and behavior is worrisome. Seizures with LGS unfortunately do not respond well to seizure medications. Behavior problems can be very challenging in many people. Psychosocial needs of people with LGS and their families are critical to address early and on an ongoing basis, because the needs of the person with epilepsy and their family change over time.
- Establish an epilepsy team early that includes epilepsy doctors, nurses, social workers, mental health providers, and rehabilitation specialists. An epilepsy center has a team of health care professionals who specialize in the diagnosis, care, and treatment of people with difficult to control seizures. Find an epilepsy center near you.
- Getting support from other families living with LGS can be invaluable. Reach out to family organizations like the LGS Foundation for support, connection, resources and services.
Summary
Lennox-Gastaut Syndrome continues to present great challenges to children and adults with the syndrome, their families, and their caregivers. Much more research is needed to identify better therapies of all types.
New advances in seizure detection, recording, and alerting, as well as other safety and protection devices, are avenues that need further attention by families, health care providers, and researchers. These types of treatments and services can improve safety management and quality of life for people with LGS and their caregivers.
- Find out what happens in a clinical trial and why it’s important to participate
- Find a clinical trial
- Learn about our efforts to advance innovation to help families in a time frame that matters. Donate now to support this important work.
- Explore the Epilepsy Pipeline Tracker for information about epilepsy therapies in various stages of development
- Check out the Device-apedia, an online searchable catalog of devices and apps being developed for epilepsy
Resources and Support
LGS Foundation improves the lives of people affected by Lennox-Gastaut syndrome through research, family support programs, events, webinars, conferences and other education.
- Learn more about LGS
- How many people live with LGS in the U.S.? Find out here.

National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health (NIH) Lennox-Gastaut syndromeGS
From Industry
- LGS Together supported by Lundbeck in the U.S.
- Living with LGS supported by Eisai, Inc.
Learn about the role of seizure alerts and download the following fact sheet to determine the best fit for you or your loved one
Find specialty care at an epilepsy center
Watch this webinar recorded in December 2014 on Lennox-Gastaut Syndrome. The speakers were Patty Osborne Shafer RN, MN; David M. Ficker MD and Angel Hernandez MD.
Learn More:
Donate to Support Our MissionResources
Epilepsy Centers
Epilepsy centers provide you with a team of specialists to help you diagnose your epilepsy and explore treatment options.
Epilepsy Medication
Find in-depth information on anti-seizure medications so you know what to ask your doctor.
Epilepsy and Seizures 24/7 Helpline
Call our Epilepsy and Seizures 24/7 Helpline and talk with an epilepsy information specialist or submit a question online.
Tools & Resources
Get information, tips, and more to help you manage your epilepsy.
Related Stories

Press Releases
Epilepsy Foundation Partners with Iaso Ventures to Launch the NeuroImpact Fund
Read Story